Scientists from the University of Tartu developed a calculation method which helps quickly identify the genes causing drug resistance in bacteria. The new method would enable a more accurate way to determine the characteristics of the bacteria causing infection in patients and thereby make the treatment more personalised.
A bioinformatics professor at the University of Tartu, Maido Remm, leads the work group that developed this new calculation method. The scientists call the method PhenotypeSeeker and it allows to quickly find the genes of bacteria inducing drug resistance.
The scientists tested the method with finding the causes for the drug resistance of the bacteria Pseudomonas aeruginosa and Clostridium difficile. The new method could be used in labs for a more accurate determination of the characteristics of bacteria causing infection, and establish precision medicine through this, i.e., provide people with a more personalised treatment with the help of genetic data.
Where do superbacteria come from?
The drug resistance of bacteria is an ever-growing problem in medicine as well as in veterinary medicine. The drug resistant bacteria are sometimes figuratively called superbacteria as they do not respond to common antibiotics or other drugs.
Drug resistance often occurs in places where an abundance of antibiotics is used. One of these fields is animal husbandry, where animals are given antibiotics in order to prevent getting ill, but where bacteria are released into the environment at the same time, which favours their development into being drug resistant. Another similar place is hospitals, which have a lot of disease-causing bacteria and where an immense amount of antibiotics is used.
Due to all of this, it becomes increasingly important for the infection-causing bacteria to be described before starting treatment. This way, a more accurate treatment can be prescribed and an excessive use of antibiotics is avoided.
One possibility for describing the bacterium as accurately as possible is to determine the drug resistance of the studied bacterial strains from their genome sequences. This is where the calculation method and software PhenotypeSeeker created by UT scientists can be used.
What does the PhenotypeSeeker do?
When bacterial strains are divided into two groups that can be called “good” and “bad”, the PhenotypeSeeker can be used to quickly find which genes are different between these two groups. In other words, the model helps to figure out which genes make the “bad” bacterial strains “bad”.
When this model is compiled for all of the main active agents of antibiotics, this model set can be used to quickly predict resistance in the studied bacterial strain. The calculation of the method for each strain takes only a few seconds. The prerequisite for the calculations is the presence of the genome sequences of the studied bacterial strain, but hopefully the DNA sequencing service will become more and more available in the coming years.
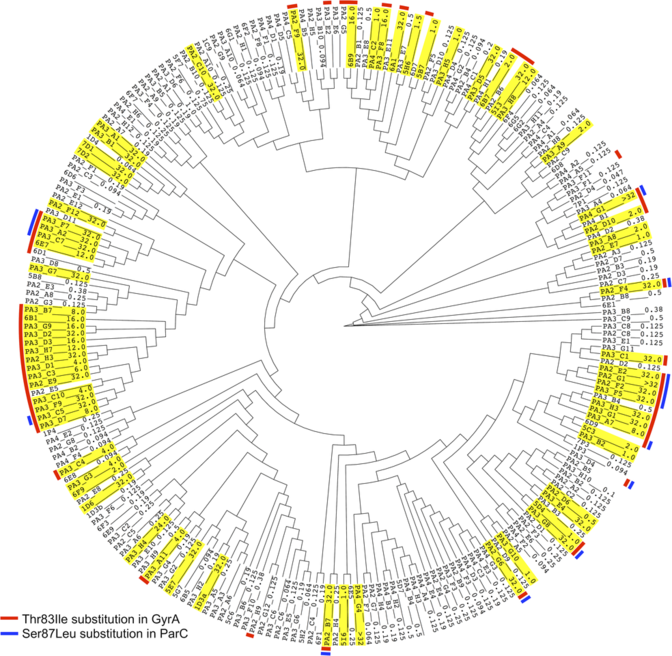
Author: Aun et. al. 2018
The use of genome sequences in medical microbiology is spreading more and more widely.
This calculation method is based on short DNA sections called k‑mers or oligomers taken from the genome sequence. When the oligomers characteristic to the “bad” bacteria are found, they can be used for building a statistical model which predicts, even in the case of bacterial strains that have not been studied yet, whether they belong to the “bad” or rather to the “good” group.
At the beginning of November, an article describing the PhenotypeSeeker was published in the top journal of the field, PLoS Computational Biology. The article described the computational side of the methodology and tested the methodology for finding the reasons behind the drug resistance of the bacteria Pseudomonas aeruginosa and Clostridium difficile.
In this research, scientists also modelled the genetic specifities of the invasive strains of the widely spread Klebsiella pneumoniae. The created models can predict the characteristics of the bacterial strains with an accuracy of 88–97 per cent.
The original part of the method is the use of oligomers in building a statistical model.
Using the PhenotypeSeeker, it is possible to retrieve the more significant oligomers from the model and later study the genes further related to resistance and the biological mechanisms which give the bacterial strain certain characteristics.
This is of interest to molecular biologists and geneticists who want to get a clearer understanding of the reasons behind the development of resistance. In addition to predicting resistance, the method is also suitable for studying and predicting the genetic background of the characteristics of interest in bacteria.
Remm added that the method has great potential in prescribing a more accurate treatment in microbiology. The k-mer based DNA analysis methods will also find application in fetal DNA analysis from mother’s blood sample.
The main developer of the methodology is the doctoral student in the bioinformatics work group, Erki Aun, supervised by Age Brauer and Maido Remm. A close cooperation with the professor of antimicrobial compounds, Tanel Tenson, and his work group was a great help. Tanel Tenson and Veljo Kisand led the sequencing of the genomes of Pseudomonades used in the research.
The translation of this article from Estonian Public Broadcasting science news portal Novaator was funded by the European Regional Development Fund through Estonian Research Council.



